



2Department of Genetics, Afyonkarahisar Health Science University, Afyonkarahisar, Turkey DOI : 10.26663/cts.2020.0002
Summary
Background: Pectus excavatum (PE) is one of the most common chest wall deformations. However, the pathogenesis of the disease is not completely understood and the results of the researches remain inconsistent. There are not enough studies in the literature investigating the most common anomalies accompanying PE. Thus, in this study, we aimed to evaluate the prenatal, natal, postnatal, genetic and clinical findings associated with congenital PE.Materials and Methods: Eighteen patients with PE (10 males, 8 females) who admits Afyonkarahisar Health Sciences University were included in the study between 2012 and 2018. Patients were investigated clinically, radiologically and genetically to determine the etiology and risk factors of PE. PE is accompanied by several anomalies and has many systemic effects. The ages of the study patients who applied to the dysmorphology clinic and included in the study ranged between 0 and 18. The findings and genetic results of approximately 120 piece dysmorphological parameters of prenatal, natal and postnatal periods of PE patients were investigated.
Results: Five parameters associated with PE were recorded in this study, which include intrauterine growth retardation (IUGR), high Beighton score, hypotonia, cryptorchidism and microcephaly.
Conclusions: Treatment of PE requires a detailed and multidisciplinary approach.
Introduction
In a patient with PE, the breastbone is sunken into his/her chest. In severe cases of pectus excavatum the chest can look as if carved, leaving a deep dent. PE, also called funnel chest, is more common in boys than girls are and severe cases can eventually interfere with heart and lung functions. There are several hypotheses regarding the pathogenesis of PE. In one of the first hypotheses, Bauhinus et al., mentioned that hypertension of the diaphragm during embryonic development could be a pathophysiologic factor. However, recent hypotheses focus on the defective sternocostal cartilage metabolism that results in biomechanical weakness and overgrowth of the sternocostal cartilage. A systematic histological analysis of the sternocostal cartilage of patients with PE has identified premature aging of the cartilage. An ultrastructural and biochemical study, which was designed to identify the reason for deterioration of sternal cartilage, revealed that the costal cartilage elements obtained from patients with PE were normal. The dietary zinc amount is correlated with the decreased metabolic activity of chondrocytes. Hence, in light of these findings there is a correlation between metabolic lesions and mechanical properties of sternal cartilages of patients with PE [1,2]. The most common chest wall deformities are PE and pectus carinatum which constitutes 95-97% of all chest wall deformities and some rare disorders such as cleft sternum, asphyxiating thoracic dystrophy (Jeune syndrome), pentalogy of Cantrell, spondylothoracic dysplasia (Jarcho–Levin syndrome) and Poland syndrome represent the remaining ~3–4% of the deformities [2]. PE is the most common chest wall deformity, which is caused by the dorsal deviation of the sternum and the 3rd to 7th rib, or costal cartilage that represents 90% of all chest wall deformities. Thoracic organ and spine deformities may be present depending on the severity of PE. Although in most cases, PE has little or no effect on the functions of underlying organs, the cosmetic appearance may produce psychological problems that require treatment. The main treatment of PE is by surgical intervention. There are several operative methods in the literature. There are several hypotheses regarding the pathogenesis of PE, however, the underlying mechanism is still not identified. There are questions about the role of developmental processes in the pathophysiology of PE [1].Dysmorphology is the study of human congenital malformations and birth defects, particularly those that affect the morphology of individuals. This term describes the body (such as stature, hand, feet and neck) and the face characteristics (head shape, nose length, ear position, vermillion thickness, etc.) of individuals compared to the same age group and ethnicity. A genetic etiology should be suspected if a child has one or more of these dysmorphic features: Congenital anomalies, growth retardation, underdeveloped secondary sexual characteristics or ambiguous genitalia and developmental delay and/or intellectual disability and/or developmental regression [3].
Head circumference measurement is an indirect and simple method to determine the normal progression of brain growth, which provides information about the intracranial volume. Growth of head circumference is very important for the normal development of the brain [4].
Hypotonia is an impairment, which is associated with many different conditions regarding genetic neuromuscular, connective tissue, central nervous system, connective tissue, and/or metabolic origins [5]. Muscle tone is clinically assessed by evaluating the resistance to passive stretching, and a reduction in the muscle tonus is defined as hypotonia. Stiffness of the muscles, tendons and soft tissues, as well as muscle contraction, contributes to muscle tone. Voltage reflex mechanism pathologies and the reduction of the segmental motor neuron excitability have been suggested as the physiological etiology of hypotonia [5].
Cryptorchidism is the absence of one or both testes within the scrotum. The incidence of cryptorchidism is high and nearly 3% of boys are undergoing an operation for this disorder in the western countries. Cryptorchidism results from the abnormalities of the hypothalamo-pituitary-testicular axis it is present in almost all of the individuals with one testis and abnormal sexual characteristics [6].
The presence of a history of hypoxic birth is important. Hence, the APGAR score (calculated from the parameters of activity, pulse, grimace, appearance and respiration) of the patients is recorded. These patients are followed for a long term in the medical genetic clinics, particularly for the presence of hypotonia. The patients are classified in one of 3 categories according to the APGAR score; low (0-3), intermediate (4-6) or normal (7-10). A low APGAR score at birth is associated with increased risk of neurological diseases, such as cerebral palsy, cognitive disorders or epilepsy [7,8]. We aimed to evaluate the genetic and clinical findings associated with congenital PE. In this way, we tried to determine the risk factors accompanying PE.
A scoring method was developed by Carter and Wilkinson in 1964 to determine generalized joint hypermobility. Later in 1973, this scoring method was modified by Beighton et al. Beighton Scoring, which we use today to determine generalized joint hypermobility, was defined. In this scoring system, the total scoring is between 0 and 9. If the total score is 5 or more, it is considered generalized joint hypermobility [9].
Methods
Written informed consent was obtained from all participants and their families and the study was performed under a protocol approved by the Afyon Kocatepe University Medical Ethics Committee (2018/9).Eighteen patients (10 males and 8 females) were enrolled in the study between 2012 and 2018. The hospital records of the study patients were investigated for the presence of the anomalies related to PE. The hospital records of the patients were investigated from the prenatal period and fetal characteristics such as fetal movements, polyhydramnios, oligohydramnios, intrauterine growth retardation (IUGR) and the maternal diseases such as diabetes, hypertension, and preeclampsia were recorded.
Besides, the birth style, birth weight and time of birth were recorded and the reason for caesarian section was investigated if it was preferred as the birth style.
Statistical Analysis
The statistical analysis was performed by Statistical Package for Social Sciences version 18.0 (SPSS Inc., SPSS IBM, Armonk, NY, USA). Continuous data were expressed as mean ± standard deviation while categorical data were expressed as numbers or percentages where appropriate. Chi-square test was used for the statistical comparison of two groups and a p-value less than 0.05 was accepted as statistically significant.
Results
The most common comorbidities were observed in patients with PE. IUGR was present in 4 out of 18 patients in the prenatal period. A diagnosis of IUGR was established at the mean 20th week of the pregnancy. The average birth weight of the study patients was 1980 gr.In 3 patients, the 5th postpartum minute APGAR score was between 4-7 (mild hypoxic birth trauma). Two of the patients was and APGAR score of less than 4 (severe hypoxic-ischemic birth trauma). These patients were followed in the neonatal intensive care unit for the long term.
The head circumference of the study patients was recorded for 2 years after birth. Microcephaly was present in 3 of the patients at birth. The average head circumference of these 3 patients was 33 cm and these patients remained to have microcephaly when they were 2 years old.
Hypotonia was present in 2 of the study patients and cryptorchidism was detected in 2 of the 10 male patients both of which were bilateral.
It was also identified in our study that the mean Beighton scores were elevated. Six of the patients had high Beighton scores and the Beighton score of all patients was 5 or more.
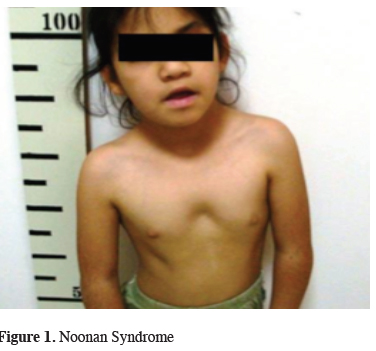
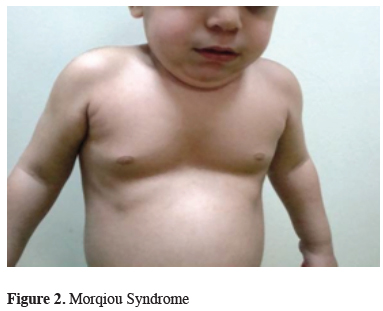
The genetic tests revealed that; 2 patients had Marfan Syndrome, 2 patients had Ehler Danlos Syndrome type 1, 2 patient had Turner Syndrome, 2 patients had Rasopathies (1 BRAF mutation and 1 SOS1 mutation) (Figure 1), one patient had Fragile X syndrome, one patient had Sotos syndrome, one patient had Digeorge Syndrome and one patient had mucopolysaccharidosis (MPS) type 4a (Morqiuo Syndrome) (Figure 2).
){kind=link}
){kind=link}
 Click Here to Zoom |
Figure 1: Noonan Syndrome |
 Click Here to Zoom |
Figure 2: Morqiou Syndrome |
Dysmorphıc Facıal Features are shown in Table 1 and the clinical and genetic results of the patients are shown in Table 2.
){kind=link}
){kind=link}
Discussion
It was previously reported that patients with PE have biochemical and histopathological abnormalities in the costal cartilage and the patients affected from PE show varying degrees of loose connective tissue [7]. In accordance with that, all of our study patients had genetic syndromes that affect the skeletal system.There was no similar article in the literature investigating our clinical data. Therefore, it is difficult to compare our results with the literature.
Marfan Syndrome and Marfanoid properties were present in approximately 15% of the patients. It is known that Marfan Syndrome is associated with pectus excavatum, scoliosis, aortic dilatation and lens dislocation. Patients with Marfan Syndrome had more severe PE. Marfan Syndrome was present in two of our patients.
In their study, Nuss et al., reported two sisters and a younger brother with severe PE. The frequency of familial pectus excavatum is 40% [2]. In our study, 5 of the patients had a definite genetic etiology.
In cases with mucopolysaccharidosis (MPS), PE is observed due to “dysostosis multiplex” which is a quite specific radiological expression. In most of the MPS disorders, skeletal abnormalities are early and prominent features and the degree of skeletal involvement varies between MPS subtypes. Most patients have radiographic dysostosis multiplex, which includes PE, abnormally shaped vertebrae and ribs, spatulate ribs, enlarged skull, bullet-shaped metacarpals, hypoplastic epiphyses, and thickened diaphyses. Thoracolumbar kyphosis or gibbus deformity are often present and important in the diagnosis [8].
In conclusion, the cases of PE show genetic and clinical heterogeneity as seen in our study. Therefore, the approach to the patients should begin with a detailed anamnesis. Genetic inquiry and research should be done in detail. Parental consanguinity marriage and the presence of PE in the family members should be questioned. Afterward, a detailed physical examination and radiological imaging should be performed. The approach to the patients should be multidisciplinary and holistic. With the development of technology, diagnosis of molecular diseases has become easier. However, the importance of obtaining a detailed anamnesis and clinical examination is still important. Therefore, no matter how far technology progresses, the multidisciplinary and holistic approach to the patients will always be important. Multidisciplinary evaluation is very important in rare diseases such as PE.
Declaration of conflicting interests
The authors declared no conflicts of interest with respect to the authorship and/or publication of this article.
Funding
The authors received no financial support for the research and/or authorship of this article.
Reference
1) Brochhausen C, Turial S, Müller FK, Schmitt VH, Coerdt W, Wihlm JM, et al. Pectus excavatum: history, hypotheses and treatment options. Interact Cardiovasc Thorac Surg 2021;14:801-6.
2) Goretsky MJ, Kelly RE, Croitoru D, Nuss D. Chest wall anomalies: pectus excavatum and pectus carinatum. Adolesc Med Clin 2004;15:455-71.
3) Allanson JE, Biesecker LG, Carey JC, Hennekam RCM. Elements of morphology: introduction. Am J Med Genet 2009; 149A: 2-5.
4) Miles JH, Hadden LL, Takahashi TN, Hillman RE. Head circumference is an independent clinical finding associated with autism. Am J Med Genet 2000;95:339-50.
5) Haller Jr JA, Scherer LR, Turner CS, Colombani PM. Evolving management of pectus excavatum based on a single institutional experience of 664 patients. Ann Surg 1989;209:578.
6) Cortes D. Cryptorchidism-aspects of pathogenesis, histology and treatment. Scand J Urol Nephrol 1998:196:1-54.
7) Tocchioni F, Ghionzoli M, Messineo A, Romagnoli P. Pectus excavatum and heritable disorders of the connective tissue. Pediatr Rep 2013;5:e15.