



2Department of Pathology, University of Health Sciences, Ankara Atatürk Chest Diseases and Thoracic Surgery Training and Research Hospital, Ankara, Turkey DOI : 10.26663/cts.2021.0009
Summary
Background: Ewing sarcoma (ES) is an uncommon aggressive malignant tumor of the bone and/or soft tissue and belongs to peripheral primitive neuroectodermal tumor (PNET) family of tumors. Bone PNETs primarily arise from the diaphysis of long bones, where as soft tissue PNETs are most commonly found in the chest wall. Multidisciplinary treatments, including chemotherapy, surgery, radiotherapy, or all three combined, improve the survival of patients with localized ES. However, the best approach to achieve local control remains controversial.Materials and Methods: We retrospectively analysed the medical records and pathology data of 14 patients (8 male, 6 female; mean age, 23.2 [range, 4-54] years) with primary thoracic ES who underwent surgery in our clinic between January 2002 and December 2019. In addition, the treatment modalities and tumor-related factors of chest wall ES and lung parenchyma were evaluated.
Results: The most frequent complaint was chest pain (n = 7). In 10 patients, the tumor originated from the ribs, whereas the remaining 4 patients had lung parenchymal tumors. Ten patients underwent complete tumor excision with chest wall resection, one patient underwent lower lobectomy with chest wall resection, and three patients underwent complete tumor excision via wedge resection. All patients were treated with chemotherapy, except two who underwent bone marrow transplantation. The median follow-up was 31.6 (range, 2-84) months. Relapses were seen in 5 cases in the median 19.8th (range, 4-60) month.
Conclusions: Complete tumor resection is the most effective treatment for thoracic ES and multimodal therapy (surgical resection, chemotherapy, and local radiation therapy), which is recommended when indicated, constitutes the optimal treatment for ES. Although relapses occur within the early postoperative period, late relapses are not uncommon. The follow-up periods must be short and should be maintained long term for late relapses.
Introduction
Ewing sarcoma is an aggressive malignancy with a high incidence of local recurrence and distant metastasis, which are important prognostic factors. Surgical resection, chemotherapy, and local radiation therapy, when indicated, constitutes the optimal treatment. Thoracic sarcomas frequently involve the ribs, characterized by indolent progression. Hereby, these tumors are often diagnosed at a locally advanced stage, with massive pleural cavity involvement, or even at a metastatic stage (in 25% of cases) [1]. Although the relapses occur within the early postoperative period, late relapses are not uncommon. The follow-up periods must be short and should be maintained long term for late relapses.ES primarily affects the bones and/or soft tissues and mainly occurs in children and young adults. Approximately 10% of ES arises from the chest wall [2]. Although ES can develop in any bone or soft tissue, the most common sites are flat bones or long bones of the leg, such as the pelvis, axial skeleton, and femur. Extraskeletal ES is by far less common than skeletal ES. ES located in the duodenum, neck, and kidney was reported in some case series. However, primary ES arising from the lung is extremely rare [3]. Along with malignant small-cell tumors of the thoracopulmonary region (Askin tumor), primitive neuroectodermal tumor (PNET), extraosseous ES (EES), and atypical ES, and these malignancies are part of a spectrum of neoplastic diseases known as the ES family of tumors. These tumors were considered to be derived from a common cell of origin due to their similar histopathological and immunohistochemical characteristics and shared non-random chromosomal translocations [4].
The patients have metastasis at diagnosis about 20%–30%, and the presence of metastasis at the time of diagnosis is the most unfavorable prognostic factor. The treatment of chest wall ES involves intense local therapy with surgical resection and/or radiation therapy, and chemotherapy is used to treat systemic diseases. In addition to surgery, chemotherapy improves the outcome from 15%-20% to 60%-70% [5]. In this study, we retrospectively analysed the findings of the cases with primary thoracic ES, and total resection surgery was performed.
Methods
We retrospectively analysed the medical records and pathological data of 14 patients (8 male, 6 female; mean age, 23.2 [range, 4-54] years) with primary thoracic ES who underwent surgery in Ataturk Chest Disease and Thoracic Surgery Training and Research Hospital, Thoracic Surgery Department between January 2002 and December 2019. The pathological material was reviewed by an experienced sarcoma pathologist. The presence of small round cell tumors with CD99 positivity without lymphoma, rhabdomyosarcoma, or neuroblastoma pathological features was assessed as ES. A panel of immunohistochemical stains, including CD99, synaptophysin, chromogranin, neuron-specific enolase, S-100, vimentin, leukocyte common antigen, cytokeratins (AE1/AE3), desmin, and actin, was used.Data of patients who underwent surgical resection in our clinic were evaluated retrospectively for age, gender, clinical features, tumor size and localization, clinical-stage, surgical procedure, resection margins, chemotherapy, radiotherapy, and treatment results. Informed consent was obtained from all patients. Approval was obtained from Ataturk Chest Disease and Thoracic Surgery Training and Research Hospital of the local institutional review board (No: 03.12.2020/ 703).
All cases were staged with clinical history, physical examination, routine laboratory tests, chest radiograph, and computed tomography (CT) of the chest and brain. Positron emission tomography (PET)–CT was used for staging in three cases, while others were evaluated using additional abdominal ultrasound for metastasis. Bronchoscopy and transthoracic needle biopsy with CT were applied in all patients preoperatively.
In this study, all patients had primary thoracic ES and underwent complete resection with safe margins. Event-free survival (EFS) was assessed using the date of disease progression, relapse, death, or last follow-up visit from the date of diagnosis. The overall survival was calculated from the date of diagnosis to the date of death or last follow-up.
Thirteen patients with Thoracic ES were diagnosed using tomography-assisted transthoracic biopsy (n = 3), incision biopsy (n = 3), metastasectomy (n = 5), supraclavicular lymph node biopsy (n = 1), and endobronchial cryobiopsy (n = 1); however, these patients were excluded from the study due to distant metastasis or unresectable/inoperable tumor. Patients with isolated thoracic ES who underwent total resection were included in the study.
Results
The most frequent complaint was chest pain (n = 7, 50%), followed by cough (n = 4, 28.5%), swelling of the chest wall (n = 2, 14.2%), and hemoptysis (n = 1, 7.1%). Table 1 shows the clinical features and demographics of patients. All patients were evaluated using chest CT. PET/CT was used for five patients. Three of the patients with PET scan were screened preoperatively and median fluorodeoxyglucose (FDG) uptake was 4.03 (2.5-6.4). A 19-year-old male who was operated on due to post-traumatic pleural hematoma and ES was detected during surgery, and a 4-year-old female patient was evaluated using PET/CT in the postoperative period for staging; both these patients showed no evidence of distant metastasis.){kind=link}
Table 1: General characteristics of patients who were operated for thoracic Ewing sarcoma.
In 10 patients, the tumor originated from the ribs, whereas the remaining 4 patients had lung parenchymal tumors. Surgical procedures included complete tumor excision with chest wall resection (n = 10), lower lobectomy with chest wall resection (n = 1), and complete tumor excision via wedge resection (n = 3). All the patients were treated with chemotherapy, except for two patients who underwent bone marrow transplantation. The median follow-up was 31.6 (range, 2-84) months. Relapse was seen in 5 cases (35.7%) in the median 19.8th (range, 4-60) month (Table 2).
){kind=link}
ES was located on the right side in 9 cases (64.2%). The mean tumor size was 10.4 (range, 2-25) cm. All cases showed diffuse membranous staining only for CD99.
A 14-year-old male patient had a 2-cm lymph node on the superior to the pancreas. Abdominal ultrasound and magnetic resonance imaging were used to determine the nodule, and histopathological signification could not be done due to the localization of the nodule; however, no progression was seen on the 48th month follow-up period.
The endobronchial lesion was determined in a 20-year-old female patient via bronchoscopy, and a biopsy was performed (Figure 1). Three cycles of neoadjuvant chemotherapy were administered to the patient.
){kind=link}
 Click Here to Zoom |
Figure 1: Thorax CT showing a Ewing sarcoma in a 20-year-old female patient at the right lower lobe (Right lower lobectomy and 9th rib resection was performed). |
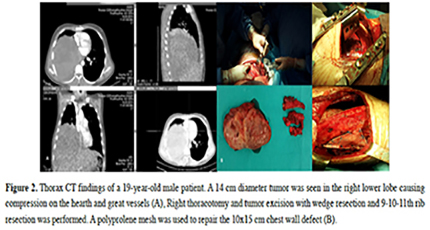
A 19-year-old male patient was admitted to the hospital due to chest pain, 45 days following a traffic accident. Chest x-ray revealed pleural effusion and tube insertion was performed due to hemothorax. Thorax CT revealed pleural effusion and a 14x12 cm heterogeneous solid lesion, suggesting a suspicious hematoma. Following this, a right thoracotomy was performed confirming the tumor. The tumor was totally excised with wedge resection of the 9-11th ribs. Postoperative PET/CT was used to determine distant metastasis and no abnormal FDG uptake was found (Figure 2). Surgical procedures included complete tumor excision with chest wall resection (n = 10), lower lobectomy with chest wall resection (n = 1), and complete tumor excision via wedge resection (n = 3).
){kind=link}
 Click Here to Zoom |
Figure 2: Thorax CT findings of a 19-year-old male patient. A 14 cm diameter tumor was seen in the right lower lobe causing compression on the hearth and great vessels (A), Right thoracotomy and tumor excision with wedge resection and 9-10-11th rib resection was performed. A polyprolene mesh was used to repair the 10x15 cm chest wall defect (B). |
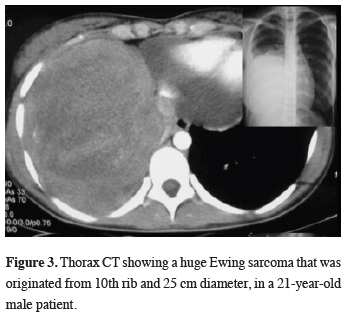
A 21-year-old male patient had a giant tumor measuring 25 cm adjacent to the right lower lobe (Figure 3). Total tumor excision and 9-10-11th rib excision was performed. However, the recurrence was occurred on the 16th month and he died on the 18th month of the follow-up.
){kind=link}
 Click Here to Zoom |
Figure 3: Thorax CT showing a huge Ewing sarcoma that was originated from 10th rib and 25 cm diameter, in a 21-year-old male patient. |
The 10 patients with ES originating from the ribs underwent complete tumor excision with chest wall resection, one patient underwent lower lobectomy with chest wall resection, and 3 patients without chest wall invasion underwent complete tumor excision via wedge resection. All patients were treated with chemotherapy postoperatively. Three cycles of neoadjuvant chemotherapy were used for a 20-year-old female patient (Table 1, case #4). Multi-drug chemotherapy, which is a minimum of four drugs such as vincristine, doxorubicin, dactinomycin, and cyclophosphamide, was used for all patients, except for two patients who underwent bone marrow transplantation. Radiotherapy was used for two patients due to narrow surgical margins and large tumor sizes.
Relapses were seen in 5 patients: 4 male and 1 female with a median age of 26.4 (range, 20-39) years. The average tumor size was 10.4 cm (range, 2-25 cm).The clinical features and treatment modalities are shown in Table 1. Chemotherapy was used in all patients, except for one patient who underwent additional bone marrow transplantation. The median follow-up was 31.6 (range, 2-84) months. Relapses were seen in 5 cases in the median 19.8th (range, 4-60) months. Three of them underwent secondary thoracic surgery procedures (redo-thoracotomy metastasectomy, contralateral wedge resection, and decortication). A 20-year-old female patient who underwent redo-thoracotomy and a 21-year-old male patient with a 25-cm chest wall tumor died on the 16th and 18th months, respectively, after diagnosis with a mortality rate of 14.3%.
Discussion
In this study, we report the clinical features, treatment strategies and outcomes, complications, and survival of 14 patients with thoracic ES who underwent surgical resection. In addition, we found that there was a male predilection. ES mainly occurs in children and young adults. Reportedly, ES is the 3rd most common primary malignant neoplasm of bone and soft tissues seen in children after osteosarcoma and rhabdomyosarcoma, respectively [6]. Extraskeletal ES/peripheral PNETs (pPNETs) are usually present in the 2nd decade of life [6]. Familial cases have been reported, and Extraskeletal ES/pPNET may rarely occur as a 2nd malignant neoplasm [7,8].The most common sites for ES are flat bones or long bones of the leg, mostly located in the pelvis, axial skeleton, and femur. Extraskeletal ES incidence is variable and ranges from 1/5,000000 to 1/10,000000. In our study, ES developed or invaded the ribs in 8 patients (72.7%), whereas 27.3% of them were extraskeletal. The typical clinical presentation for ES is pain or swelling around the tumor site [8]. In this study, cough was a more frequent complaint than swelling.
EES/pPNET is often large at presentation, ranging from 1 to 40 cm in diameter and frequently larger than 10 cm [6]. In this study, the mean tumor size of extraskeletal and skeletal ES was 6.3 (2-12) cm and 13.6 (5-25) cm, respectively.
Classic ES/pPNET has patterns of formless sheets and lobules of uniform round to oval cells, and spindle cell areas may be present [9]. Mitoses are present in variable numbers and are inconspicuous in some cases [6]. The overlapping histologic, immuno-histochemical, and cytogenetic, and molecular genetics features cause diagnostic challenges despite significant clinical and prognostic differences. EES and PNETs are pathologically characterized by diffuse sheets of small round cells that express vimentin as well as CD99 (a MIC2 gene product). The diagnosis can be confirmed by the non-random chromosomal translocation involving the ES breakpoint region 1 gene; t (11;22) (q24;q12) [10].
A capsule or pseudo-capsule frequently appears in the resected tumor. In some cases, it was difficult to identify any residual gross tumor in the margin, which is important for the decision of radiotherapy [11,12]. Positive or close margins are generally managed by radiotherapy. In this study, radiotherapy was used for 2 patients with 13 and 25 cm ES on the chest wall due to close surgical margins. A 21-year-old male patient with a 25-cm tumor had a recurrence on the 16th month of the follow-up and eventually died on the 18th month (case # 9). Furthermore, a 14-year-old male patient with a 13-cm tumor had a 48-month EFS postoperatively.
There are limited studies concerning the management of primary chest wall ES. The treatment of ES is multimodal, which includes definitive surgery, radiation therapy, or a combination of both, followed by induction chemotherapy. Radiation therapy is applicable in unresectable tumors or those that prove to have margins of resection involved or closely approached by a tumor. Treatment condensation for high-risk patients may consist of high-dose chemotherapy, followed by stem cell transplant. In this study, 2 male patients aged 13 and 27 with 8 and 2-cm tumor sizes, respectively, underwent bone marrow transplantation [6,13,14]. The 27-year-old patient had a recurrence in the 60th month following left lower lobectomy, and wedge resection for the right lower lobe was performed. Both the patients were followed up for 36 and 84 months, respectively without mortality.
Prognosis is associated with age, localization, metastasis, tumor size, origin tissue of tumor, chemotherapy administered, and response to chemotherapy [15,16]. The current 5-year EFS rate for non-metastatic EWS/pPNET ranges from 60%–75% [17].
Surgical resection of chest wall ES can be performed as the initial therapy (up-front) or after the patient has received neoadjuvant chemotherapy (delayed). Delayed surgery can provide smaller resections and better margin negativity rates compared to up-front surgery [18]. Besides reducing the tumor volume and increasing the rate of negative margin resections (R0), the other benefit of neoadjuvant chemotherapy is decreasing the use of radiotherapy [1]. A study of 98 patients with chest wall ES showed that patients who underwent delayed surgery had a higher frequency of successful complete resection of the tumor and decreased requirement of radiation therapy to the chest [19]. The primary aim is to provide complete resection with adequate margins. Neoadjuvant chemotherapy prevents the patient from radiation therapy and its potential long-term complications, including pulmonary fibrosis, increased incidence of coronary artery disease, and a significant incidence of secondary tumors [19]. Radiation therapy in doses recommended for the treatment of ES/PNET has been associated with a significant incidence of secondary tumors in these patients (10%–30%), and delayed resection resulted in a significant decrease in the proportion of patients requiring chest radiation therapy [19]. However, in this study, only one patient received neoadjuvant chemotherapy and relapse occurred in the 4th month postoperatively.
The main limitation of this study was its retrospective nature and the benefit of total resection of the affected ribs could not be proved.
In conclusion, although ES is an aggressive disease with a high incidence of local recurrence and distant metastases, long-term survival is achievable with appropriate and aggressive multimodal therapy. Relapses are not rare, but if they occur, surgical resection for secondary tumors can help improve survival. Surgical treatment with adequate margins in association with appropriate use of combined chemotherapy and postoperative radiotherapy or bone marrow transplantation, when indicated, is necessary for preventing relapses and achieving longer survival rates. Further prospective and larger sample size studies are recommended to determine the prognostic factors and treatment results of thoracic ES.
Declaration of conflicting interests
The authors declared no conflicts of interest with respect to the authorship and/or publication of this article.
Funding
The authors received no financial support for the research and/or authorship of this article.
Ethics approval
The study was approved by the institutional review board of Health Sciences University, Ataturk Chest Disease and Thoracic Surgery Training and Research Hospital (No: 03.12.2020/ 703).
Authors’ contributions
SH, YA; co-wrote the paper, collected the data, performed the analysis, KA, PB; contributed data/analysis tools, GF; conceived and designed the analysis, co-wrote the paper, SŞEG, MŞD; collected the data, SK; conceived and designed the analysis.
Reference
1) Provost B, Missenard G, Pricopi C, Mercier 3, Mussot S, Fabre D et al. Ewing Sarcoma of the Chest Wall: Prognostic Factors of Multimodal Therapy Including En Bloc Resection. Ann Thorac Surg 2018; 106: 207-13.
2) Denbo J, Orr WS, Wu Y, Wu J, Billups CA, Navid F et al. Timing of Surgery and the Role of Adjuvant Radiation Therapy in Ewing Sarcoma of the Chest Wall: A Single Institution Experience. Ann Surg Oncol 2012; 19: 3809-15.
3) Hancorn K, Sharma A, Shackcloth M. Primary extraskeletal Ewing’s sarcoma of the lung. Interact Cardiovasc Thorac Surg 2010; 10; 803-4.
4) Jedlicka P. Ewing Sarcoma, an enigmatic malignancy of likely progenitor cell origin, driven by transcription factor oncogenic fusions. Int J Clin Exp Pathol 2010; 19: 338-47.
5) Seker MM, Kos T, Ozdemir N, Seker A, Aksoy S, Uncu D et al. Treatment and Outcomes of Ewing Sarcoma in Turkish Adults: A Single Centre Experience. Asian Pac J Cancer Prev 2014; 15: 327-30.
6) Tsokos M, Alaggio RD, Dehner LP, Dickman PS. Ewing sarcoma/peripheral primitive neuroectodermal tumor and related tumors. Pediatr Dev Pathol 2012; 15: 108-26.
7) Zoubek A, Simonitsch I, Panzer-Grümayer ER, Ghali D, Pfleiderer C, Haas OA et al. Ewing tumor after treatment of Ki-1+ anaplastic large cell lymphoma: therapy associated secondary neoplasm or unrelated coincidence? Cancer Genet Cytogenet 1995; 83: 5-11.
8) Spunt SL, Rodriguez-Galindo C, Fuller CE, Harper J, Krasin MJ, Billups CA et al. Ewing sarcoma family tumors that arise after treatment of primary childhood cancer. Cancer 2006; 107: 201-6.
9) Cavazzana AO, Ninfo V, Roberts J, Triche TJ. Peripheral neuroepithelioma: a light microscopic, immunocytochemical, and ultrastructural study. Mod Pathol 1992; 5: 71-8.
10) Holcombe EG. The Ewing Family of Tumors: Ewing’s Sarcoma and Primitive Neuroectodermal Tumors. Pediatr Clin North Am 1997; 44: 991-1004.
11) Takahashi D, Nagayama J, Nagatoshi Y, Nishiyama M, Yokoyama R, Moriyasu Y et al. Primary Ewing’s sarcoma family tumors of the lung a case report and review of the literature. Jpn J Clin Oncol 2007; 37: 874 -7.
12) Schuck A, Hofmann J, Rübe C, Hillmann A, Ahrens S, Paulussen M et al. Radiotherapy in Ewing’s sarcoma and PNET of the chest wall: results of the trials CESS 81, CESS 86 and EICESS 92. Int J Radiat Oncol Biol Phys 1998; 42: 1001-6.
13) Castex MP, Rubie H, Stevens MCG, Gauzy JS, Brouchet AG, Rey A et al. Extraosseous localized Ewing tumours: improved outcome with anthracyclines. The French Society of Pediatric Oncology and International Society of Pediatric Oncology. J Clin Oncol 2007; 25: 1176-82.
14) .Granowetter L, Womer R, Devidas M, Krailo M, Wang C, Bernstein M et al. Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children’s Oncology Group Study. J Clin Oncol 2009; 27: 2536-41.
15) Denbo JW, Orr WS, Wu Y, Wu J, Billups CA, Navid F et al. Timing of surgery and the role of adjuvant radiotherapy in ewing sarcoma of the chest wall: a single-institution experience. Ann Surg Oncol 2012; 19: 3809-15.
16) Lin PP, Jaffe N, Herzog CE, Costelloe CM, Deavers MT, Kelly JS et al. Chemotherapy response is an important predictor of local recurrence in Ewing sarcoma. Cancer 2007; 109: 603-11.
17) Womer RB, West DC, Krailo MD, Dickman PS, Pawel B. Randomized comparison of every-two-week v. every-three-week chemotherapy in Ewing sarcoma family tumors (ESFT). J Clin Oncol 2008; 26: 10504.