



2Department of Thoracic and Cardiovascular Surgery, Edgardo Rebagliati Martins National Hospital, Lima, Peru
3Service of Pathological Anatomy, Edgardo Rebagliati Martins National Hospital, Lima, Peru DOI : 10.26663/cts.2023.0027
Summary
Lung carcinoid tumors account for 1-2% of malignant lung neoplasms. Early diagnosis is crucial, and surgery at an early stage may improve the clinical picture and may even be a curative option. We present a 78-year-old woman with a history of poorly controlled arterial hypertension, anxious syndrome, irritable bowel syndrome, and paroxysmal palpitations for the last two years. Radiologic imaging showed a left hilar nodular lesion causing endobronchial obstruction in the left lower lobe. Anatomopathology revealed a low-grade pulmonary neuroendocrine tumor, and pulmonary resection with nodal staging was promptly considered. The diagnosis of pulmonary carcinoid tumor is challenging; therefore, it is necessary to maintain suspicion in patients with non-specific and/or persistent respiratory symptoms, and complete resection and prolonged close follow-up should be considered despite a satisfactory postoperative course.Introduction
Pulmonary carcinoid tumors are a group of neuroendocrine tumors originating from Kulchitsky neuroendocrine cells [1] and account for 1-2% of malignant lung neoplasms, with an incidence of 1.49/1,000,000 population [2]. They occur in all age groups with a peak incidence around the fifth decade. The clinical presentation is silent in up to 25- 39% of cases, being diagnosed with non-specific symptoms, or with cough (15%), hemoptysis (24%), dyspnea (22%), recurrent pneumonia, atelectasis (7%), and rarely as carcinoid syndrome (8%) [3]. They are classified into typical carcinoid (TC), atypical carcinoid (AC), large cell neuroendocrine carcinoma (LCNC)/small cell lung carcinoma (SCLC) (1), considered analogous to grade 1, 2, and 3 neuroendocrine tumors of other locations respectively, being according to the mitotic index and necrosis present in typical (< 2 mitoses/2 mm2 with the absence of necrosis and more than 0.5 cm) and atypical (2-10 mitoses/mm2 or necrosis often punctate). Diagnosis is based on imaging studies to identify the presence of hilar involvement, endobronchial tumors, signs of bronchial obstruction, mediastinal lymph node involvement, and pulmonary nodules. Some reviews have described that the main location is at the segmental or subsegmental bronchi level (80%), and others mention that up to 75% of cases present endobronchial lesions [4]. This latter presentation makes the use of flexible bronchoscopic techniques necessary as the best method for the anatomopathological study. Surgery is the main treatment of choice, and surgical indication at an early stage can mean clinical improvement and even a definitive curative option, especially in typical carcinoma, which is why it is important to know about this entity.Case Presentation
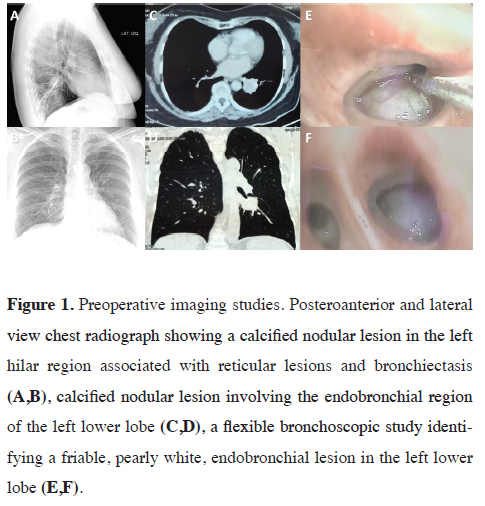
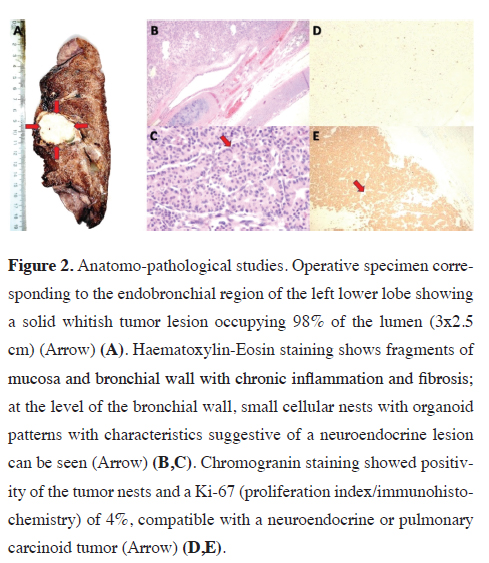
A 78-year-old woman with a history of uncontrolled arterial hypertension, irritable bowel syndrome, anxiety syndrome, and paroxysmal palpitations for two years. She received symptomatic medical treatment and with the passage of time, liquid stools, abdominal pain, and persistent precordial pain were added, for which reason she went for medical consultation. A sinus tachycardia with supraventricular extrasystoles was identified and a chest X-ray showed a nodular image in the left para-retrocardiac region. The tomographic study showed a left lesion of 3x3 cm, of solid characteristics with involvement of the left lower lobe bronchus (LLB), and due to the high suspicion of malignancy, she underwent a bronchoscopic study (Figure 1). This last evaluation reported a friable endobronchial lesion at the level of the LII entry that caused an occlusion of 90% of the bronchial lumen, and finally, the anatomopathological study revealed small cellular nests with organoid patterns with characteristics suggestive of the neuroendocrine lesion at the bronchial wall level. Immunohistochemistry was compatible with a low-grade (Grade I) pulmonary neuroendocrine tumor in bronchial mucosa TTF-1: (Negative), Chromogranin: (+++), CK20: (Negative), PanCK: (+), Ki67: (2%). After diagnostic confirmation, the tumor was resected, which was included next to the left lower lobe, and the corresponding lymph node resection was performed. The study of the operative specimen also showed involvement of the submucosa and part of the bronchial cartilage, with a mitotic index (< 1%) and Ki67 index: (4%) (Figure 2). The patient had an optimal postoperative recovery and was discharged with the indication for periodic outpatient follow-up. After nine months, no signs of recurrence have been observed and the patient has shown significant clinical improvement, with no additional medication and good functional class. Written informed consent was obtained from the patient for the publication of her data.){kind=link}
){kind=link}
 Click Here to Zoom |
Figure 1: Preoperative imaging studies. Posteroanterior and lateral view chest radiograph showing a calcified nodular lesion in the left hilar region associated with reticular lesions and bronchiectasis (A,B), calcified nodular lesion involving the endobronchial region of the left lower lobe (C,D), a flexible bronchoscopic study identifying a friable, pearly white, endobronchial lesion in the left lower lobe (E,F). |
 Click Here to Zoom |
Figure 2: Anatomo-pathological studies. Operative specimen corresponding to the endobronchial region of the left lower lobe showing a solid whitish tumor lesion occupying 98% of the lumen (3x2.5 cm) (Arrow) (A). Haematoxylin-Eosin staining shows fragments of mucosa and bronchial wall with chronic inflammation and fibrosis; at the level of the bronchial wall, small cellular nests with organoid patterns with characteristics suggestive of a neuroendocrine lesion can be seen (Arrow) (B,C). Chromogranin staining showed positivity of the tumor nests and a Ki-67 (proliferation index/immunohistochemistry) of 4%, compatible with a neuroendocrine or pulmonary carcinoid tumor (Arrow) (D,E). |
Discussion
Carcinoid tumors can produce vasoactive hormones, whose activity rarely conditions the appearance of carcinoid syndrome (8%) and its crises (3%) as reported by Savu et al, who described this presentation in 3 out of 98 patients, characterized by tachycardia, arterial hypertension, flushing, bronchospasm and accelerated digestive transit [5]. Differential diagnoses include asthma, airway obstruction by aspirated foreign body, metastases, bronchopulmonary carcinoma, benign pulmonary nodule, or endocrinological entities such as Cushing's syndrome, which can occur in up to 2 % of AC and TC due to ectopic production of adrenocorticotropic hormone (ACTH) or acromegaly, due to ectopic production of growth hormone-releasing hormone (GHRH) or insulin-like growth factor 1 (IGF-1). ACTH, growth hormone (GH) testing is not routinely performed unless there are related symptoms.They are usually visualized as solitary pulmonary nodules predominantly in segmental or subsegmental bronchi (80%), in some cases with calcifications, and in up to 75% they are associated with endobronchial lesions as in the present case [4]. Histopathology of TC and AC consist of a uniform organoid growth pattern with cytological features consisting of a moderate amount of eosinophilic cytoplasm with an eosinophilic matrix, with a variety of histological patterns in both, including spindle cell, trabecular, palisade, glandular, follicular, rosette, pink, clear cell, and papillary patterns. Identification and classification can be difficult with immunohistochemistry being important, which points towards a neuroendocrine nature by being positive for synaptophysin, chromogranin, and CD56. However, these can be positive in 30% of cases of lung adenocarcinoma and squamous cell carcinoma, as well as metastatic carcinomas of the breast, prostate, and other sites, and should therefore be considered as differential diagnoses. TTF-1 expression in TC and AC is varied. Most carcinoids stain for cytokeratins, but up to 20% to 25% may be keratin negative. Ki-67 staining shows a low proliferation rate in TC, usually less than 5% while in AC it is higher, usually between 5% and 20%. The proliferation rate may be more useful in small biopsies to separate TC and AC from SCLC, respectively. Ki67 is of limited value in tumors of lung origin, however, it has been estimated that a value > 5% in AC and > 10%, in general, are associated with poor prognosis [6]. The transcription factors TTF-1, CDX-2, and PDX-1 allow differentiation of primary lung origin, being site-specific for lung, gastrointestinal and pancreatic origins, respectively.
Approximately 80 % of low-grade (typical) and 60 % of intermediate-grade (atypical) pulmonary carcinoids express somatostatin receptors by immunohistochemistry and can be imaged using positron emission tomography (PET) or somatostatin receptor scans (OctreoScan) [7]. Typically, 90% are confined to the bronchi and 10% to regional lymph nodes; however, in our case, the nodal staging was negative. As with any neoplastic process, prognosis depends on histological type, stage, and metastatic involvement. Surgical management is of choice and even curative in some cases, there is no standardization, with complete anatomical resection plus mediastinal lymphadenectomy being the most commonly described, however, options range from wedge resection, lobectomy, pneumonectomy or sleeve resection together with mediastinal lymph node study. The latter is controversial, due to the low potential for nodal metastasis of non-active AC and TC described. However, in endocrinologically active tumors a high nodal metastatic potential is presumed, as demonstrated by Seastedt et al who after evaluating 68 cases of pulmonary carcinoids associated with Cushing's syndrome found mediastinal nodal involvement in 37%, with an overall incidence of persistence/recurrence of 16.2% and with a median time to recurrence of 55 months (Range, 18-152 months). He also reported a disease-free time of 12.7 years [8].
Girelli et al analyzed 325 patients with pulmonary carcinoids undergoing complete resection (236 with lymph node involvement and 89 without lymph node involvement), of these 23.6% had TC, 39.3% AC, and 37.1 % large cell neuroendocrine carcinoma. 58.4 % underwent lobectomy, and 21.3 % pneumonectomy, with a mean tumor size of 11-130 mm. Mortality analyzed after 4.0 years of follow-up reached 5.1 %. For TC and AC, the 5-year survival was 89 % and 78 % at 10 years, with a worse prognosis for large cell neuroendocrine carcinoma, 47 % at 5 years, and 41 % at 10 years [9]. Follow-up is carried out taking into account the high potential for recurrence, by clinical and radiological examination. At 3 and 6 months and depending on whether it is TC or AC, annually and semi-annually respectively. Bronchoscopic control may even be indicated in the case of endoscopic resections. These measures should be maintained even for life, with a 10-year survival rate of approximately 90%. The patient underwent follow-ups at 3, 6, and 9 months with no evidence of recurrence [10]. Somatostatin receptor (SRS)-based therapies such as 177-Lu-DOTATATE is an effective and safe option in patients with somatostatin receptor (SRS)-positive (TC)- and (AC)-positive progression. Zidan et al reported in 48 patients after 42 months of follow-up a 33% mortality, a progression-free survival of 23 months, and an overall survival of 59 months [7]. In cases where surgical management is not possible, other therapies such as local radiotherapy should be considered and combined with surgery in cases of AC and Large Cell Neuroendocrine Carcinoma.
In conclusion, it should be remembered that the prognosis and survival of these patients also depend on the histological type of clinical stage, with surgery being the treatment of choice even in locally advanced stages.
Declaration of conflicting interests
The authors declared no conflicts of interest with respect
to the authorship and/or publication of this article.
Funding
The authors received no financial support.
Ethical approval
The present article is approved for the committee ethical
(iO56-94/22) of Department Thoracic Surgery.
Authors' contributions
All authors contributed equally to the idea, data
collection, drafting and final approval of the manuscript.
Reference
1) Reuling EMBP, Dickhoff C, Plaisier PW, Bonjer HJ, Daniels
JMA. Endobronchial and surgical treatment of pulmonary carcinoid
tumors: A systematic literature review. J Lung Cancer
2019; 134: 85-95.
2) González-Flores E, Serrano R, Sevilla I, Viúdez A, Barriuso J,
Benavent M, et al. SEOM clinical guidelines for the diagnosis
and treatment of gastroenteropancreatic and bronchial neuroendocrine
neoplasms (NENs). Clin Transl Oncol 2019; 21: 55-63.
3) Jang S, Schmitz JJ, Atwell TD, Welch TL, Welch BT, Hobday
TJ, et al. Percutaneous Image-Guided Core Needle Biopsy
of Neuroendocrine Tumors: How Common Is Intraprocedural
Carcinoid Crisis? J Vas Interv Radiol 2021; 32: 745-51.
4) Mindaye ET, Tesfaye GK. Bronchial carcinoid tumor: A case
report. Int J Surg Case Rep 2020; 77: 349-52.
5) Savu C, Melinte A, Lukadi JL, Mirvald C, Savu C, Belu E et al.
Neuroendocrine syndrome in bronchial carcinoid tumors. Exp
Ther Med 2020; 20: 200.
6) Rekhtman N. Lung neuroendocrine neoplasms: recent progress
and persistent challenges. Mod Pathol 2022; 35: 36-50.
7) Zidan L, Iravani A, Oleinikov K, Ben-Haim S, Gross DJ, Meirovitz
A et al. Efficacy and Safety of 177Lu-DOTATATE in
Lung Neuroendocrine Tumors: A Bicenter study. J Nucl Med
2022; 63: 218-25.
8) Seastedt KP, Alyateem GA, Pittala K, Steinberg SM, Schrump
DS, Nieman LK et al. Characterization of Outcomes by Surgical
Management of Lung Neuroendocrine Tumors Associated With
Cushing Syndrome. JAMA Netw Open 2021; 4: e2124739.