



2Department of Thoracic Surgery, Bozok University Medical Faculty, Yozgat, Turkey
3Department of Chest Disease, Ankara Medicalpark Hospital, Ankara, Turkey
4Department of Radiodiagnostic, Ankara Medicalpark Hospital, Ankara, Turkey DOI : 10.26663/cts.2018.00015
Summary
Inflammatory myofibroblastic tumor (IMT) is a rare thoracic lesion, the diagnosis of which should be supported by histological and immunochemical examinations. Although this is known as a benign tumor, it should be completely resected, and patients should be closely monitored to avoid any local invasion or recurrence. This article presents a 34-year-old female patient who has endobronchial polypoid IMT that is rarely seen among IMT patients. Inferior lobectomy and lymph node sampling was performed. The patient had no local recurrence and metastasis free at the end of first year postoperatively and follow-up is ongoing.Introduction
Inflammatory myofibroblastic tumor (IMT) is uncommon among primary lung cancers. It constitutes 0.04% to 1.2% of all lung tumors, and may occur at any age group [1]. The aim of this case report is to identify the clinical approach to this rare tumor and highlight the key features with a review of the relevant literature.Case Presentation
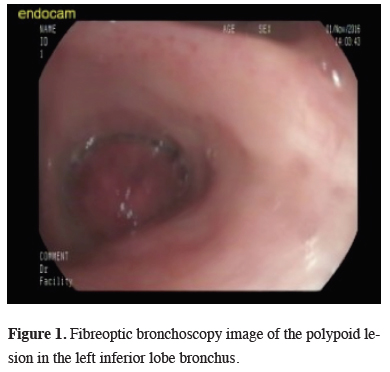
A 34-year old female presented with the complaint of a dry cough that had been ongoing for one month. On the pulmonary radiographs, a mass lesion was determined in the left inferior lobe.On the thoracic CT image, a mass lesion of 46 x 42 mm was observed with spicular extension and showing lobulation at the left inferior lobe in close proximity to the inferior pulmonary vein. On the PET-CT SUVmax value of the mass in the left inferior lobe was 20.6 and no other pathological involvement was determined. The histopathological examination of bronchoscopic biopsy reported spindle cell proliferation and inflammatory granulation tissue (Figure 1).
){kind=link}
 Click Here to Zoom |
Figure 1: Fibreoptic bronchoscopy image of the polypoid lesion in the left inferior lobe bronchus. |
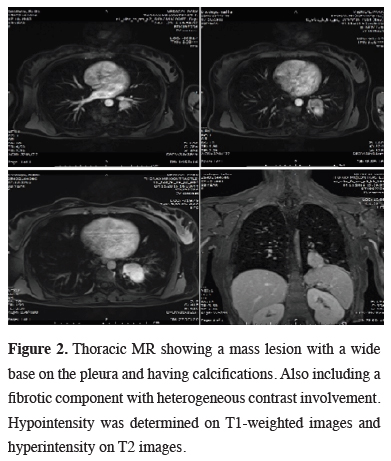
Due to the close proximity to the inferior pulmonary vein, a thoracic MR was performed, demonstrating a mass lesion with a wide base on the pleura, showing calcifications. A fibrotic component showing heterogeneous contrast involvement was seen after the administration of intravenous contrast material (Figure 2). Cranial metastasis was ruled out by cranial MR.
){kind=link}
 Click Here to Zoom |
Figure 2: Thoracic MR showing a mass lesion with a wide base on the pleura and having calcifications. Also including a fibrotic component with heterogeneous contrast involvement. Hypointensity was determined on T1-weighted images and hyperintensity on T2 images. |
A left lateral thoracotomy was performed and a biopsy was taken from the mass. The frozen section was reported as mesenchymal tumor than a lower lobectomy and lymph node sampling was performed.
The histopathologic examination reported a mesenchymal tumor in the bronchial lumen invading the bronchi and parenchyma. Tumor cells were in the form of bundles with spindle, hyperchromatic nucleus and cytoplasm with undefined borders and scattered lymphoid cells. SMA was focal positive, S-100 and CD34 was negative. The pathological diagnosis of the lesion was inflammatory myofibroblastic tumor. The surgical margins were tumor free and lymph nodes showed no metastasis.
The patient was discharged on postoperative day 5 uneventfully. At the 12-month postoperative follow-up examination, no recurrence or metastatic findings were found.
Discussion
Inflammatory myofibroblastic tumor was first reported in the lungs by Brunn in 1939 [2]. It was first named by Umiker et al. in 1954 due to the tendency to mimic a malignant process both clinically and radiologically [3]. Due to the variable cellular components, various terms have been used to describe IMT, including plasma cell granuloma, inflammatory pseudotumor, xanthogranuloma, inflammatory fibrosarcoma and pseudosarcomatous myofibroblastic proliferation [1]. In 2002, the World Health Organization classified IMT as an “intermediate malignancy (rarely metastasizing)” [4]. However, a recent study of chromosomal translocation involving ALK gene stated that it was present in 50% of IMTs, thereby suggesting that approximately 50% of IMTs could be malignant [5].IMT can develop in any age group and both genders are affected equally where the lungs and abdomen are the most commonly affected locations [5,6]. The typical location is in the lung parenchyma, but also it has been reported in the trachea or the bronchi [1,7].
Preoperative diagnosis of pulmonary IMT is difficult due to the diversity of radiological manifestations [6]. Although endobronchial polypoid forms developing in the bronchus are the most uncommon form, there have been reported cases, and chest radiography only reveals non-specific finding such as pulmonary atelectasis or obstructive pneumonia [8]. Kim et al, reported that the most common radiological presentation was a solitary, peripheral, well-circumscribed lung mass [9]. On MR imaging, the mass is typically isointense to muscle on T1-weighted images and hypo- to hyperintense on T2-weighted images, depending on the fibrous contents. Heterogeneous contrast enhancement is seen on post-contrast MR images with delayed persistent enhancement [8]. On PET scans, positive FDG uptake similar to that of a malignant tumor can be seen [1].
In the histopathological analysis, IMT can be seen to be composed of spindled myofibroblasts, fibroblasts, and inflammatory cells (lymphocytes, plasma cells, and eosinophils). It has been reported that approximately 60% of IMT cases have a rearrangement of the ALK gene on chromosome 2p23, which has been suggested as responsible for tumor transformation [5]. In immunohistochemical examinations, tumor cells show strong diffuse positivity with smooth muscle actin and vimentin, and are negative for cytokeratin, CD34 and S100. In the current case, the diagnosis of tumor was made from the general appearance and the determination of S-100 (-), and CD34 (-) [6].
There have been very few reports of multiple metastasis of IMT, and local recurrence has been reported as 6.6%-13% following incomplete resection [4,10]. For pa¬tients undergoing complete resection of IMT in the lungs, prognosis is known to be favorable with survival rates of 91% at 5 years and 77% at 10 years [10]. Of the anticancer drugs available, in some cases in which ALK is positive on immunohistochemisty staining, crizotinib, an ALK inhibitor, may be effective [7]. In the current case, this was seen with pathological examination postoperative to complete resection and no additional treatment was applied.
Declaration of conflicting interests
The authors declared no conflicts of interest with respect to the authorship and/or publication of this article.
Funding
The authors received no financial support.
Reference
1) Takeda S, Onishi Y, Kawamura T, Maeda H. Clinical spectrum of pulmonary inflammatory myofibroblastic tumor. Interact Cardiovasc Thorac Surg 2008; 7: 629 33.
2) Brunn H. Two interesting benign lung tumors of contradictory histopathology. J Thorac Surg 1939; 9: 119-31.
3) Umiker WO, Iverson L. Postinflammatory tumors of the lung; report of four cases simulating xanthoma, fibroma, or plasma cell tumor. J Thorac Surg 1954; 28: 55-63.
4) Coffin CM, Fletcher JA. Inflammatory myofibroblastic tumor. In: Fletcher CD, Unni KK, Mertens F, editors. World Health Organization classification of tumours: pathology and genet¬ics of tumours of soft tissue and bone. Lyon: IARC Press; 2002. p. 91-3.
5) Coffin CM, Patel A, Perkins S, Elenitoba-Johnson KS, Perlman E, Griffin CA. ALK1 and p80 expression and chromosomal rearrangements involving 2p23 in inflammatory myofibro-blastic tumor. Mod Pathol 2001; 14: 569-76.
6) Coffin CM, Watterson J, Priest JR, Dehner LP. Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases. Am J Surg Pathol 1995; 19: 859-72.
7) Cerfolio RJ, Allen MS, Nascimento AG, Deschamps C, Trastek VF, Miller DL, et al. Inflammatory pseudotumors of the lung. Ann Thorac Surg 1999; 67: 933-6.
8) Kim SJ, Kim WS, Cheon JE, Shin SM, Youn BJ, Kim IO, et al. Inflammatory myofibro-blastic tumors of the abdomen as mimickers of malignancy: imaging features in nine children. AJR Am J Roentgenol 2009; 193: 1419-24.