



2Department of Pathology, Istanbul University, Istanbul Medical Faculty, Istanbul, Turkey DOI : 10.26663/cts.2019.0007
Summary
Pulmonary sarcomatoid carcinoma is an extremely rare malignant lung tumor with an unfavorable prognosis, which shows carcinomatous and sarcomatous components. We present a case of 78-year-old patient with a middle lobe tumor who underwent a bronchial sleeve middle lobectomy. The tumor was a carcinosarcoma with squamous cell carcinoma and rhabdomyosarcoma components histologically. We tried to emphasize this extreme lung tumor and its clinicopathological features.Introduction
Pulmonary sarcomatoid carcinoma is an extremely rare malignant lung tumor, accounting for less than 1% of all lung tumors. It is a biphasic lung tumor with an unfavorable prognosis, which shows carcinomatous and sarcomatous components. The World Health Organization (WHO) defined five subtypes of pulmonary sarcomatoid carcinomas such as giant cell, spindle cell, and pleomorphic carcinoma, carcinosarcoma and pulmonary blastoma [1].We present a case of 78-year-old patient with a middle lobe tumor who underwent a bronchial sleeve middle lobectomy. The tumor was a carcinosarcoma with squamous cell carcinoma and rhabdomyosarcoma components histologically. We tried to emphasize this extreme lung tumor and its clinicopathological features.
Case Presentation
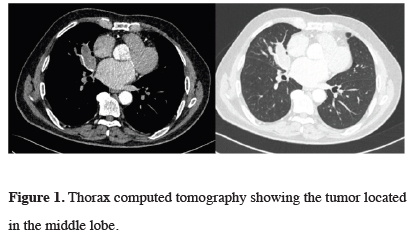
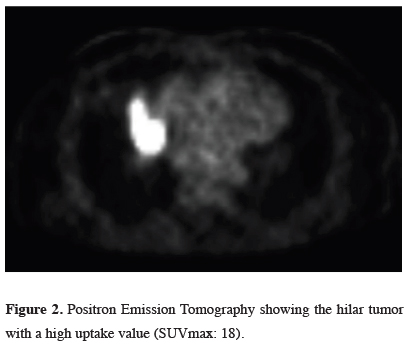
Our patient was a 78-year-old asymptomatic male who had undergone a coronary bypass surgery and was found to have a lung mass during follow- ups. He also had hypertension and a right nephrectomy for benign renal disease. He has a history of 25 pack-year smoking and asbestos exposure. A chest X - ray showed a right hilar mass appearance which was confirmed with computerized tomography. The tumor was located at the medial segment of the middle lobe at the hilar region, measuring 65x32 mm in diameter and obstructed the middle lobe bronchus (Figure 1). A whole body PET/CT showed a tumor in the middle lobe with a standard uptake value of 18 without any involvement of mediastinal lymph nodes (Figure 2).){kind=link}
){kind=link}
 Click Here to Zoom |
Figure 1: Thorax computed tomography showing the tumor located in the middle lobe. |
 Click Here to Zoom |
Figure 2: Positron Emission Tomography showing the hilar tumor with a high uptake value (SUVmax: 18). |
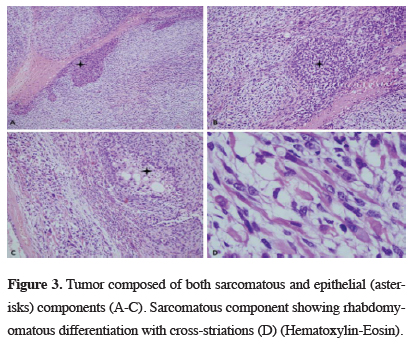
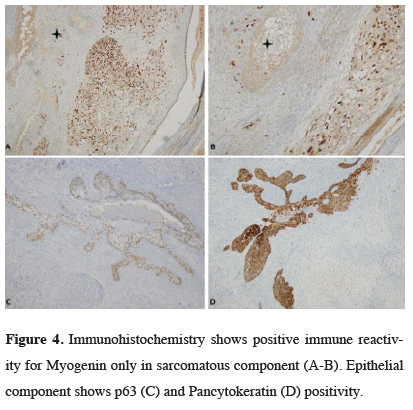
He also had two benign cystic lesions in the liver without any uptake. Cranial magnetic resonance imaging showed no metastasis. Bronchoscopy showed an endobronchial tumor at the orifice of middle lobe bronchus extending into the intermediate bronchus. Biopsy revealed necrotic tissue with a focal immature mesenchymal element consistent with either a pulmonary blastoma or other sarcomatoid carcinoma variants. No definite diagnosis could be given due to the limited tissue. The patient had no metastasis of mediastinal lymph nodes on mediastinoscopy. He underwent a right posterolateral thoracotomy and a bronchial sleeve middle lobectomy with mediastinal lymph node dissection. Histological findings showed a biphasic tumor composed mostly of spindle cell tumor with rhabdoid features. Epithelial component was focally present. Immunohistochemical examination supported the biphasic nature of the tumor. Sarcomatoid component was positive for desmin and myogenin. Epithelial component was positive for pancytokeratin, p63 and EMA (focal) (Figures 3,4).
){kind=link}
){kind=link}
 Click Here to Zoom |
Figure 3: Tumor composed of both sarcomatous and epithelial (asterisks) components (A-C). Sarcomatous component showing rhabdomyomatous differentiation with cross-striations (D) (Hematoxylin-Eosin). |
 Click Here to Zoom |
Figure 4: Immunohistochemistry shows positive immune reactivity for Myogenin only in sarcomatous component (A-B). Epithelial component shows p63 (C) and Pancytokeratin (D) positivity. |
The tumor was negative to TTF-1, SMA, CD34 and S100. Histologic diagnosis was a carcinosarcoma. A total of 80% of the tumor histologically showed features of a rhabdomyosarcoma, whereas the remainder of 20% was a squamous cell carcinoma. The patient did not receive any adjuvant chemotherapy. He is doing well at postoperative six months.
Discussion
The most recent WHO classification of lung tumors classifies the pulmonary sarcomatoid carcinomas variants into five groups such as pleomorphic, spindle cell and giant cell carcinoma, pulmonary blastoma and carcinosarcoma [1]. Pleomorphic carcinoma is a poorly differentiated carcinoma containing at least 10% spindle and/or giant cells along with a non-small cell carcinoma variant, mostly squamous cell carcinoma or an adenocarcinoma. Spindle cell carcinoma consists of only spindle cells and is identical to the spindle cell component of pleomorphic carcinoma. Giant cell carcinoma is composed entirely of highly pleomorphic giant cells. Pulmonary blastoma is a biphasic tumor with a primitive epithelial component that may resemble a well-differentiated fetal adenocarcinoma and primitive mesenchymal stroma. The last variant, pulmonary carcinosarcoma is defined as a malignant tumor with a histologically biphasic mixture of conventional non-small cell carcinoma and true sarcoma containing differentiated sarcomatous elements such as malignant cartilage, bone, or skeletal muscle [1]. The carcinomatous component in a carcinosarcoma is most often a squamous cell carcinoma as in our case, followed in frequency by adenocarcinoma and large cell carcinoma. The sarcomatous component is often poorly differentiated spindle cell carcinoma, which shows areas of more specific sarcomatous differentiation, most often rhabdomyosarcoma as seen in our case, followed by osteosarcoma or chondrosarcoma comprising combinations of these components. Tumor genotyping in pulmonary carcinosarcomas shows identical mutations in both the carcinomatous and sarcomatous components, which supports the hypothesis that both components originate from a single clone [1,2]. This finding is an evidence that sarcomatoid carcinomas represent malignant ‘‘epithelial’’ neoplasms that have undergone divergent connective tissue differentiation and they are not ‘‘collision’’ tumors [1].Pulmonary carcinosarcomas are poorly differentiated lung tumors mostly occurring in heavy smokers with an asbestos exposure at a mean age of 65 years. Patients have a male predominance with a rate of seven [3]. Similarly, our patient is a 78-year-old ex-heavy smoker with a history of asbestos exposure.
Cough, fever and hemoptysis are the most common symptoms. Chest pain, fever and malaise may also occur. A very rare symptom such as pulmonary osteoarthropathy has also been described [4]. Patients with peripheral tumors may be asymptomatic On the other hand, tumors are often located centrally with an endobronchial extension at a rate of 62%. They are frequently slow growing tumors and more likely to grow into the bronchi without any infiltration. They may show extensive invasion to the surrounding lung parenchyma. Tumor location is mostly upper lobes and the greatest dimension of the tumors has an average of 7 cm. Preoperative diagnosis with either endobronchial or transthoracic biopsy may be difficult if the biopsy specimens show only one component [5]. However, endobronchial biopsy showed biphasic histological features in our patient. Metastases to the regional lymph nodes and distant organs, particularly to the brain are common. Our patient has a big central tumor obstructing the middle lobe bronchus, however he has neither regional nor distant metastasis.
Surgery is the choice of treatment for pulmonary carcinosarcomas, however they are highly aggressive tumors with poor outcomes. Tumours with endobronchial localization have comparably better prognosis beacuse peripheral tumors tend to metastasize early and widely. The surgical outcome is not favorable with a five– year survival rate ranging from 12 to 49% [3,6,7]. Koss et al reported that the stage of the disease did not affect survival, however the tumour size was strongly related to survival. Patients with tumours less and greater than 6 cm have 40% and 10% 5-year survival rates, respectively in their study [3]. Rahuoma et al reviewed a very large national database of almost 1,000,000 patients to compare pulmonary sarcomatoid carcinomas with non-small cell lung carcinomas with respect to clinicopathological features and surgical outcomes. They reported that 4987 patients with histological diagnosis of pulmonary sarcomatoid carcinomas presented with stages that are more advanced and worse survival outcomes. They concluded that surgery provided a survival advantage for pulmonary sarcomatoid carcinomas. However, they also stated that these tumors had a higher risk of recurrence and should be evaluated for novel adjuvant therapies [8]. The sarcomatous component of pulmonary carcinosarcomas is more progressively metastatic. Thus, a chemotherapy regimen targeting the sarcomatous component should be selected. On the other hand, sarcomatoid carcinomas showed a high rate of resistance to conventional first-line chemotherapy in a series of almost 100 patients [9]. Thus, the authors proposed new therapeutic strategies based on better knowledge of the carcinogenesis of sarcomatoid carcinomas. Radiotherapy is also a well-established choice of treatment, which can reduce the local recurrence rate in cases with residual tumor [10]. Our patient is free of disease during this six- month follow-up without any adjuvant treatment.
In summary, pulmonary carcinosarcoma is a very rare lung tumor histologically classified among the sarcomatoid carcinomas. Although a definitive treatment is unclear, surgery appears to have the most favorable outcome. Adjuvant treatments might be considered for a highly possible local recurrence and distant metastasis.
Declaration of conflicting interests
The authors declared no conflicts of interest with respect to the authorship and/or publication of this article.
Funding
The authors received no financial support.
Reference
1) Corrin B, Chang YL, Rossi G, Koss MN, Geisinger K, Wick MR, et al. Sarcomatoid carcinoma. In: Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC, editors. Pathology and genetics of tumours of the lung, pleura, thymıus and heart (World Health Classification of Tumours). Lyon: IARC Press: 2004. P. 53-8.
2) Holst VA, Finkelstein S, Colby TV, Myers JL, Yousem SA. p53 and K-ras mutational genotyping in pulmonary carcinosarcoma, spindle cell carcinoma, and pulmonary blastoma: implications for histogenesis. Am J Surg Pathol 1997; 21: 801-11.
3) Koss MN, Hochholzer L, Frommelt RA. Carcinosarcomas of the lung: a clinicopathological study of 66 patients. Am J Surg Pathol 1999; 23: 1514-26.
4) Meade P, Moad J, Fellows D, Adams CW. Carcinosarcoma of the lung with hypertrophic pulmonary osteoarthropathy. AnnThorac Surg 1991; 51: 488-90.
5) Kanzaki R, Ikeda N, Okura E, Kitahara N, Okimura A, Kawahara K, Ohta M. Pulmonary carcinosarcomas with an osteosarcomatous component. Gen Thorac Cardiovasc Surg 2012; 60: 855-8.
6) Akura Y. A case of true pulmonary carcinosarcoma. Haigan Jpn J Lung Cancer 2008; 48: 191-6.
7) Petrov DB, Vlassov VI, Kalaydjiev GT, Plochev MA, Obretenov ED, Stanoev VI, Danon SE. Primary pulmonary sarcomas and carcinosarcomas-postoperative results and comparative survival analysis. Eur J Cardiothorac Surg 2003; 23: 461-6.
8) Rahouma M, Kamel M, Narula N, Nasar A, Harrison S, Lee B, et al. Pulmonary sarcomatoid carcinoma: an analysis of a rare cancer from the Surveillance, Epidemiology, and End Results database. Eur J Cardiothorac Surg 2018; 53: 828-34.